Exploiting the Principle of Locality in Electronic Structure Theory
Since the unification of molecular dynamics and density functional theory (DFT), ab initio molecular dynamics (AIMD) has become an important tool for studying dynamics of atoms in materials and molecules. Unfortunately, the computational cost of the conventional DFT grows cubically with the number of atoms, which severely limits the size of the systems that can be modeled with AIMD and, therefore, hinders its application to large heterogeneous systems (e.g. interfaces, nuclei, defects). With the growing interest in dynamical processes in nanoscale systems (e.g. biomolecules, nanomaterials, interfaces), there have been substantial efforts to develop DFT methods the cost of which grows linearly with the number of atoms. Despite substantial recent progress in this area, the cost of modern linear-scaling DFT methods remains too high to make them practical. That is why most AIMD simulations today still rely on the conventional cubically-scaling DFT and, therefore, remain limited in size and simulated time.
1. Efficient linear-scaling AIMD
Our notable first achievement in this area was to develop a new low-cost linear-scaling AIMD method for simulating large ensembles of small non-covalently bonded fragments such as molecules or ions. What makes this approach uniquely efficient and accurate is that (1) the locality of electrons, a pre-requisite for linear scaling, is ensured by approximate compact orbitals strictly localized within predefined radii around their fragments and (2) the nuclear motion is treated stochastically with a modified Langevin integrator, fine-tuned to retain stable dynamics even with approximately calculated forces. We demonstrated that the our method enables stable and reliable simulations of large collections of molecules or ions (e.g. molecular liquids and solids, ionic liquids and crystals, solutions) allowing to use AIMD to study new physical phenomena on substantially larger time and length scales (Figure 1, left).
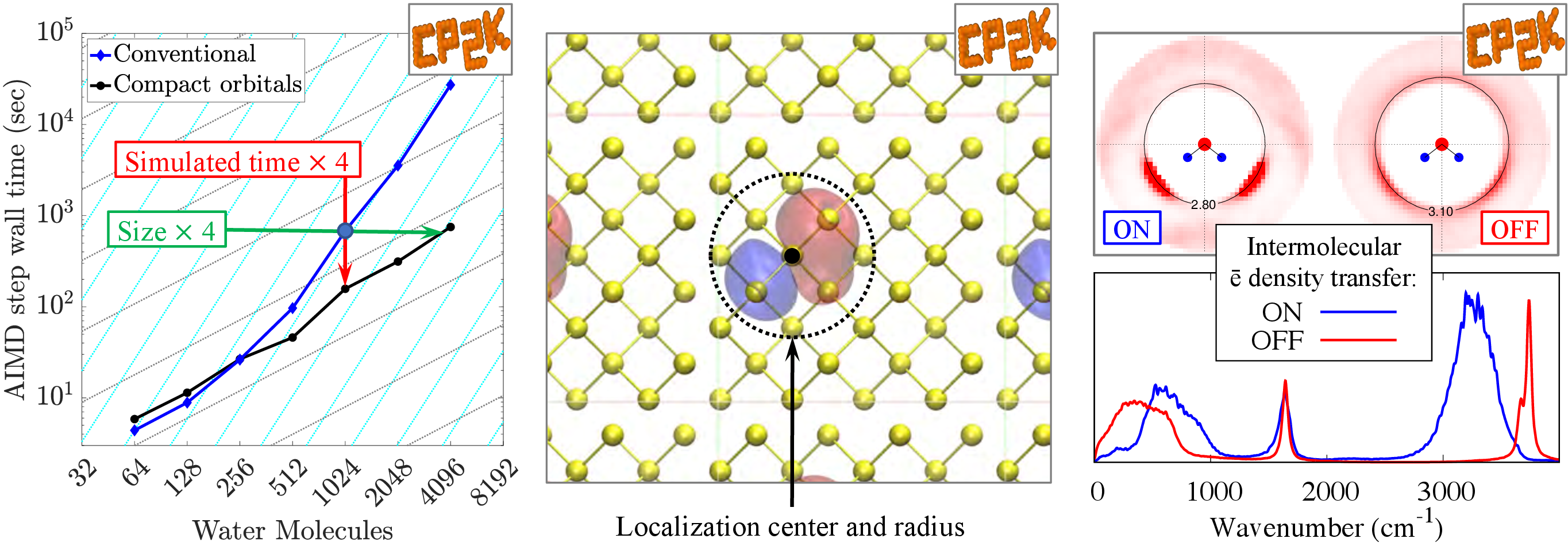
Although AIMD based on compact orbitals is overwhelmingly successful for molecular and ionic systems, the application of the same underlying DFT method to systems of covalenly bonded atoms is impossible due to a long-standing problem of finding the optimal compact representation of the electronic ground state. Guided by our breakthrough work on compact orbitals for molecular systems, we proposed a computationally efficient method to detect and obviate the optimization problems on-the-fly without introducing significant errors to the computed electronic properties. Our numerous tests showed that, despite the additional approximation, the new linear-scaling DFT method remains accurate and robust for a variety of covalent systems, opening new frontiers for AIMD studies of bond-breaking and bond-creating dynamics in nanoscale systems.
2. Electron locality for physical insight
The defining characteristic of a compact orbital (Figure 1, center) is its localization radius. This radius restricts the electron described by a compact orbital to the small region of space around orbital’s localization center. In addition to gaining substantial computational advantages, we exploited our ability to control the electron localization to obtain physical insight into the fundamental nature of chemical bonding and electronic-structure origins of observed properties of molecular systems.
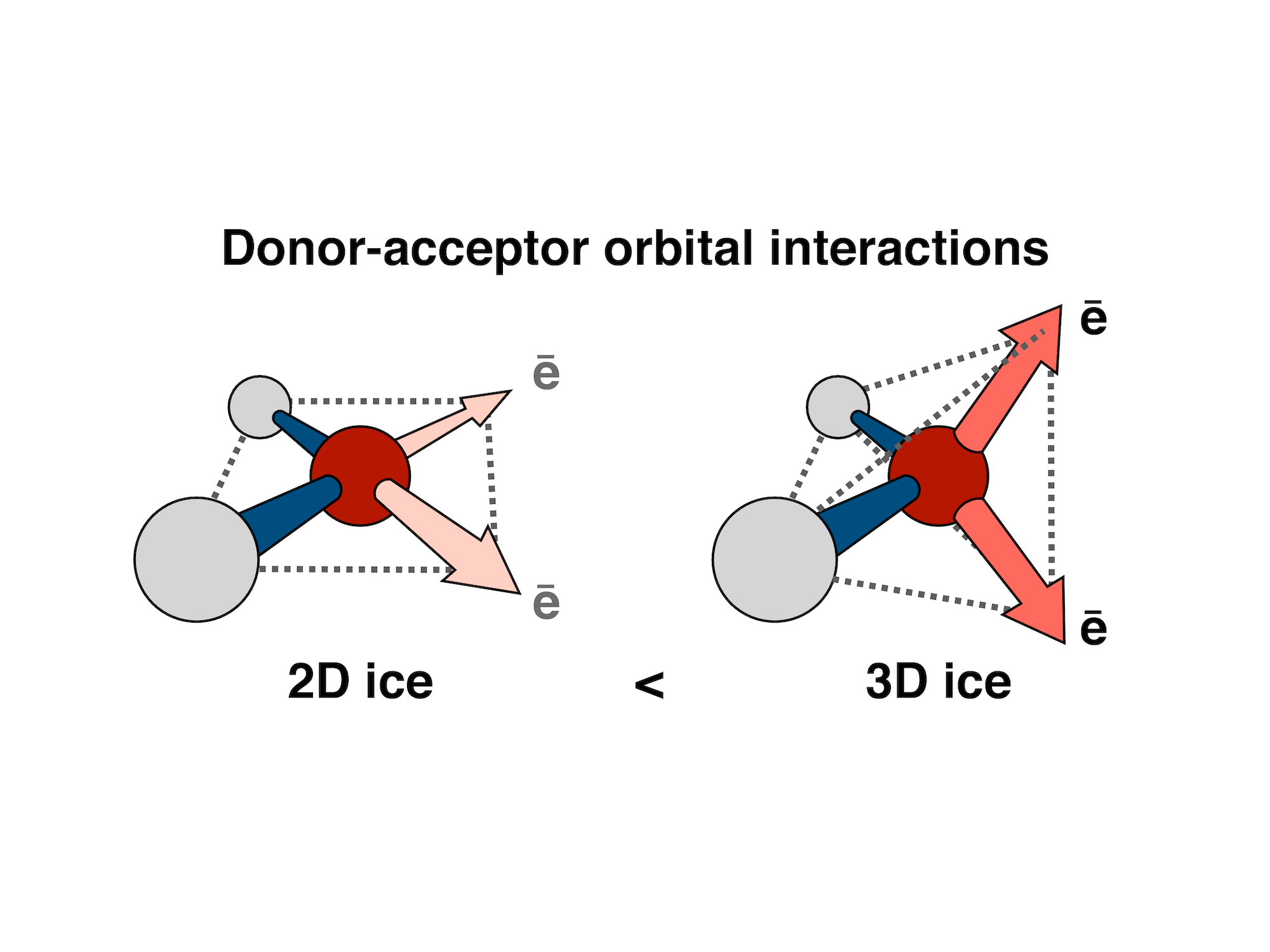
For example, we performed unprecedented AIMD simulations of liquid water with the localization radii of all compact orbitals set to zero. This trick “switched off” the electron density transfer between molecules and thus removed the donor-acceptor component from intermolecular interactions, leaving only electrostatic, polarization and dispersion forces to describe bonding between water molecules. Comparison of this intentionally unrealistic simulation to the realistic conventional simulation showed that small amount of intermolecular electron density transfer has a profound effect on the structure and dynamics of the hydrogen-bond network and, therefore, on many observable properties of liquid water (Figure 1, right). By providing unrivaled insight into the impact of hydrogen bonding on observable properties, this study aids interpretation of spectroscopic features, catalytic behavior and solvation properties of aqueous systems.
The unique ability of compact orbitals to measure the strength of pairwise donor-acceptor interactions allowed us to quantify the extent to which nanoconfinement of liquid water disrupts its hydrogen bonding and to reveal how subtle intermolecular electronic effects lead to the unusual “room-temperature ice” structure of water in confined spaces. The sensitivity of the pairwise donor-acceptor energies to small distortions in the hydrogen bond network also allowed us to measure how distortions of the nearby hydrogen bonds in liquid water are correlated in space and time, providing an important missing link between the dynamics of individual water molecules and the hotly debated large-scale structural inhomogeneity in ambient temperature liquid water.
Through collaboration, we have also been involved in the extension of our theory of intermolecular bonding analysis to systems that contain metal surfaces. This theoretical development enables deeper understanding of electronic effects driving catalytic processes on metal surfaces.
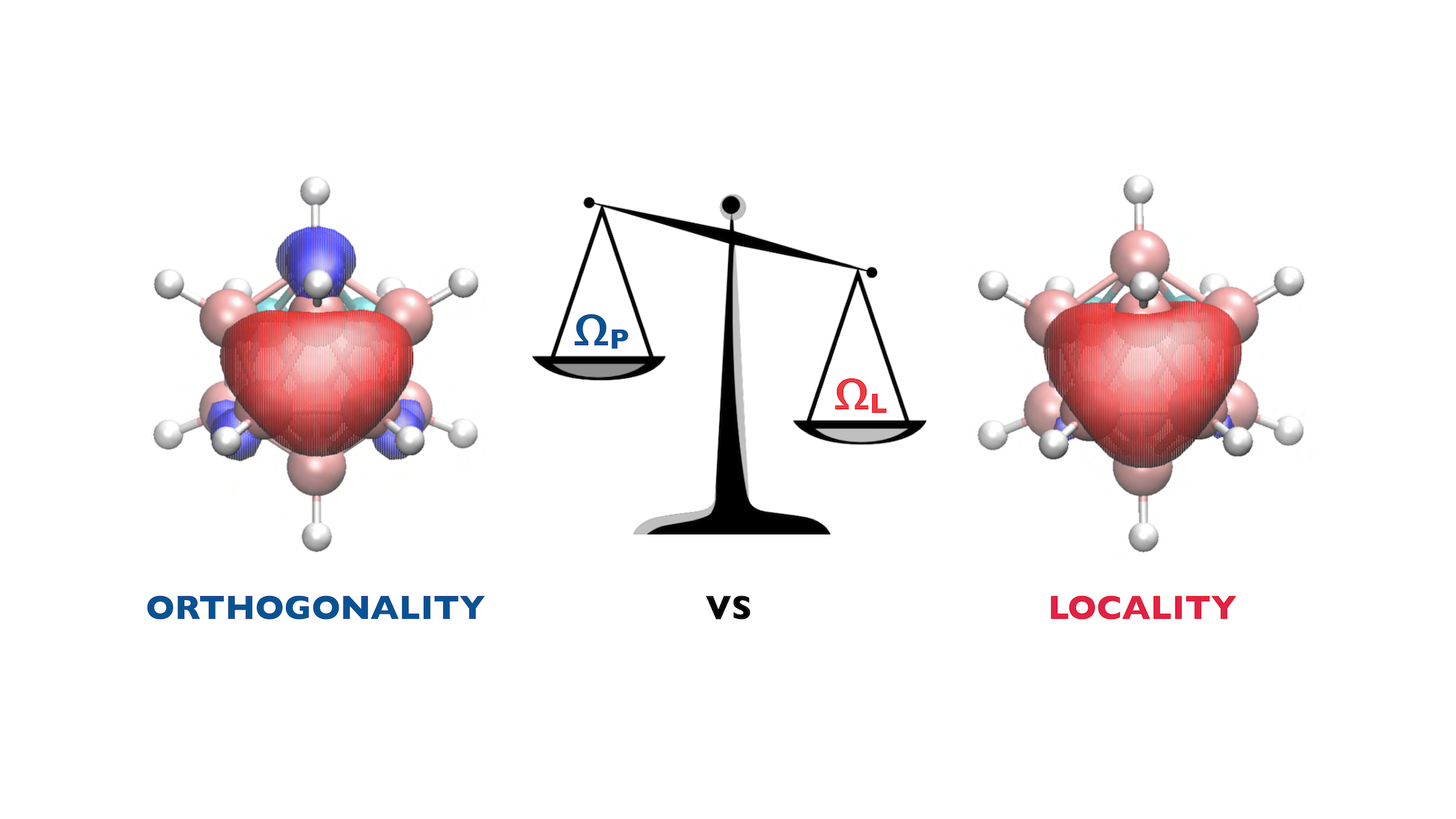
3. Variable-metric localization of molecular orbitals
Our work on compact orbitals has taken our research in an unexpected direction. In addition to constructing localized compact orbitals, we introduced a new approach to the localization of conventional molecular orbitals, which we called variable-metric localization. This approach is a generalization of existing localization methods and allows to sacrifice orthogonality of the orbitals in order to achieve better locality. After designing and testing numerous optimization algorithms, we found, to our delight, that the variable-metric method enables fast localization of not only occupied but substantially more challenging virtual orbitals, working exceptionally well for a variety of molecules and periodic materials including large systems with non-trivial bonding. The ability of the variable-metric method to improve locality of both occupied and virtual orbitals makes it promising for designing more efficient local electron correlation methods.
Variable-metric localization of occupied and virtual orbitals
The key idea of the variable-metric approach to orbital localization is to allow nonorthogonality between orbitals while, at the same…

Direct unconstrained variable-metric localization of one-electron orbitals
Spatially localized one-electron orbitals, orthogonal and non-orthogonal, are widely used in electronic structure theory to describe chemical bonding and speed…

Energy decomposition analysis for metal surface-adsorbate interactions by block localized wave functions
The energy decomposition analysis based on block localized wave functions (BLW-EDA) allows one to gain physical insight into the nature…

Robust linear-scaling optimization of compact localized orbitals in density functional theory
Locality of compact one-electron orbitals expanded strictly in terms of local subsets of basis functions can be exploited in density…
- 1
- 2