Unravelling Origins of Poorly Understood Chemical Phenomena
A significant part of our research is to use computational modeling to reveal microscopic origins of important but poorly understood chemical phenomena in bulk solid systems and on surfaces with the ultimate objective of advancing rational design of better materials or chemical processes. Many of the projects highlighted below were devised in collaboration with the colleagues in the Department of Chemistry at MgGill.
1. Mechanisms of Green Organic Reactions
A notable direction of our research is the computational investigation of mechanisms of the green organic transformations discovered or enhanced in the group of our McGill colleague Prof. Chao-Jun Li. Being complementary to inconclusive or fragmentary experimental data, our quantum chemistry modeling of molecules in the ground and excited electronic states proved useful, often crucial, in determining the microscopic mechanisms of chemical transformations in these collaborative projects.
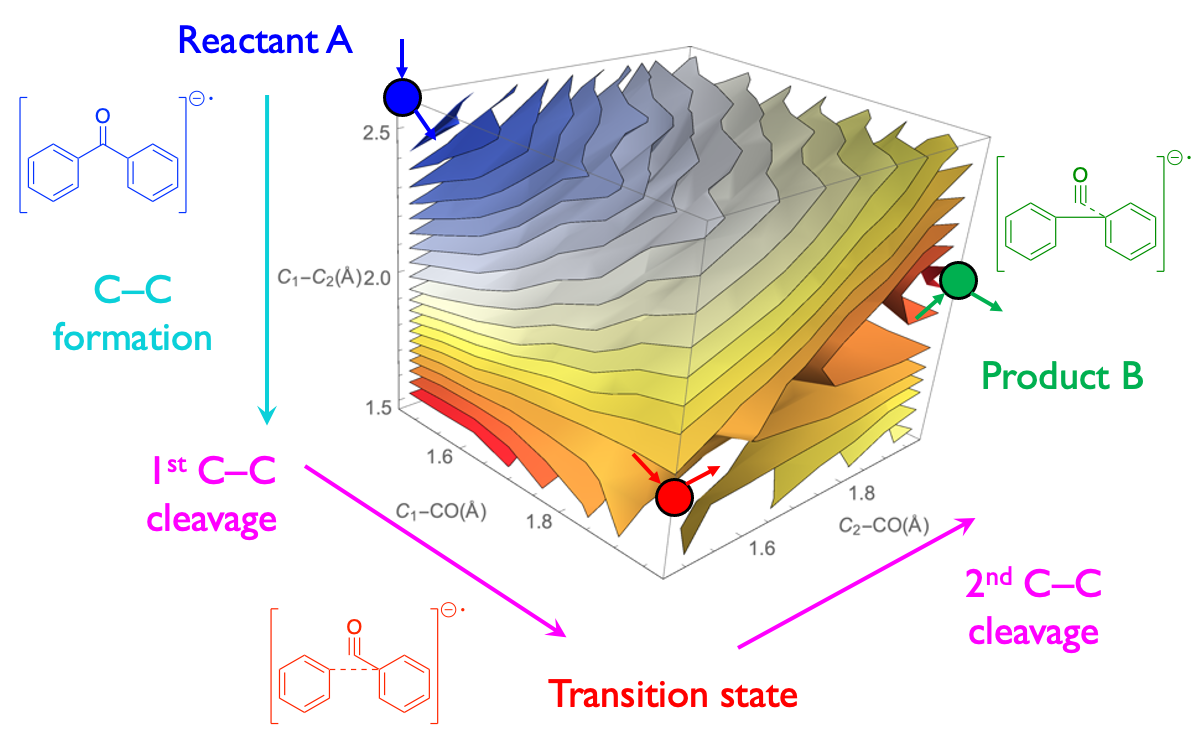
For example, it was DFT modeling that suggested that the green light-driven metal-free decarbonylation of diaryl ketones proceeds via an oxygen transfer from a solvent molecule to an excited-state intermediate — the hypothesis later supported by additional experimental measurements. Another example of practicality of DFT modeling is our systematic investigation of the mechanism of the pinacol coupling enabled by light and hydrazine that revealed a thermodynamically and kinetically plausible sequence of hydrogen atom transfer and C–C bond formation steps, which make this novel green reaction unique.
2. Dramatically different cocrystallization of similar steroids with aromatic molecules
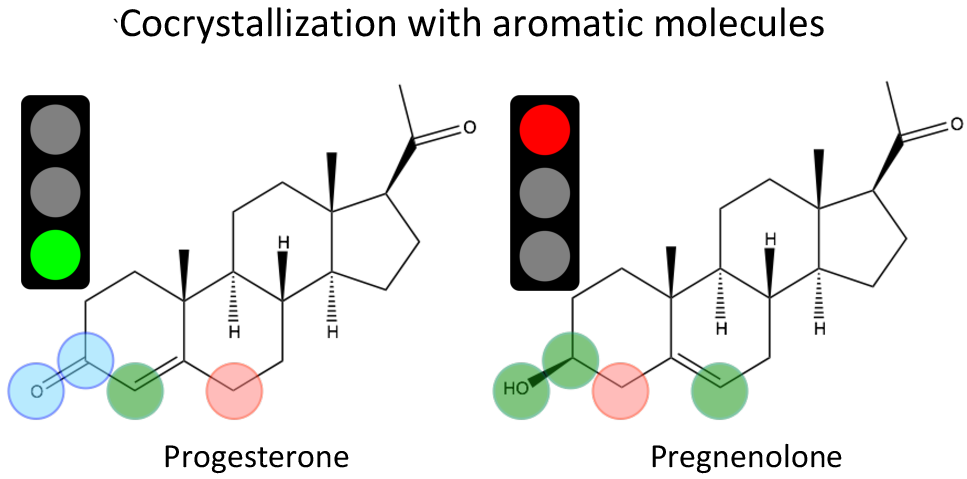
One of our challenges was to explain a puzzling observation that the steroid called progesterone forms cocrystals with a variety of aromatic molecules whereas pregnenolone (PRE), a steroid with an almost identical structure, cocrystallizes only with few. To solve this problem, we designed a simple computational procedure that generated experimentally inaccessible meta-stable cocrystals of PRE without relying on prohibitively expensive methods of crystal structure prediction. This enabled a direct comparison of intermolecular forces in the cocrystals of the two steroids and allowed us to determine that, in the case of PRE, the cocrystallization is restricted thermodynamically because of the strong hydrogen bonds in the pure PRE crystal. This fundamental analysis suggests that the design of selective biomolecular steroid receptors and steroid-specific drugs should focus on tuning the strength of hydrogen bonding in these systems.
3. Strong and reversible absorption of carbon dioxide in green metal-organic frameworks
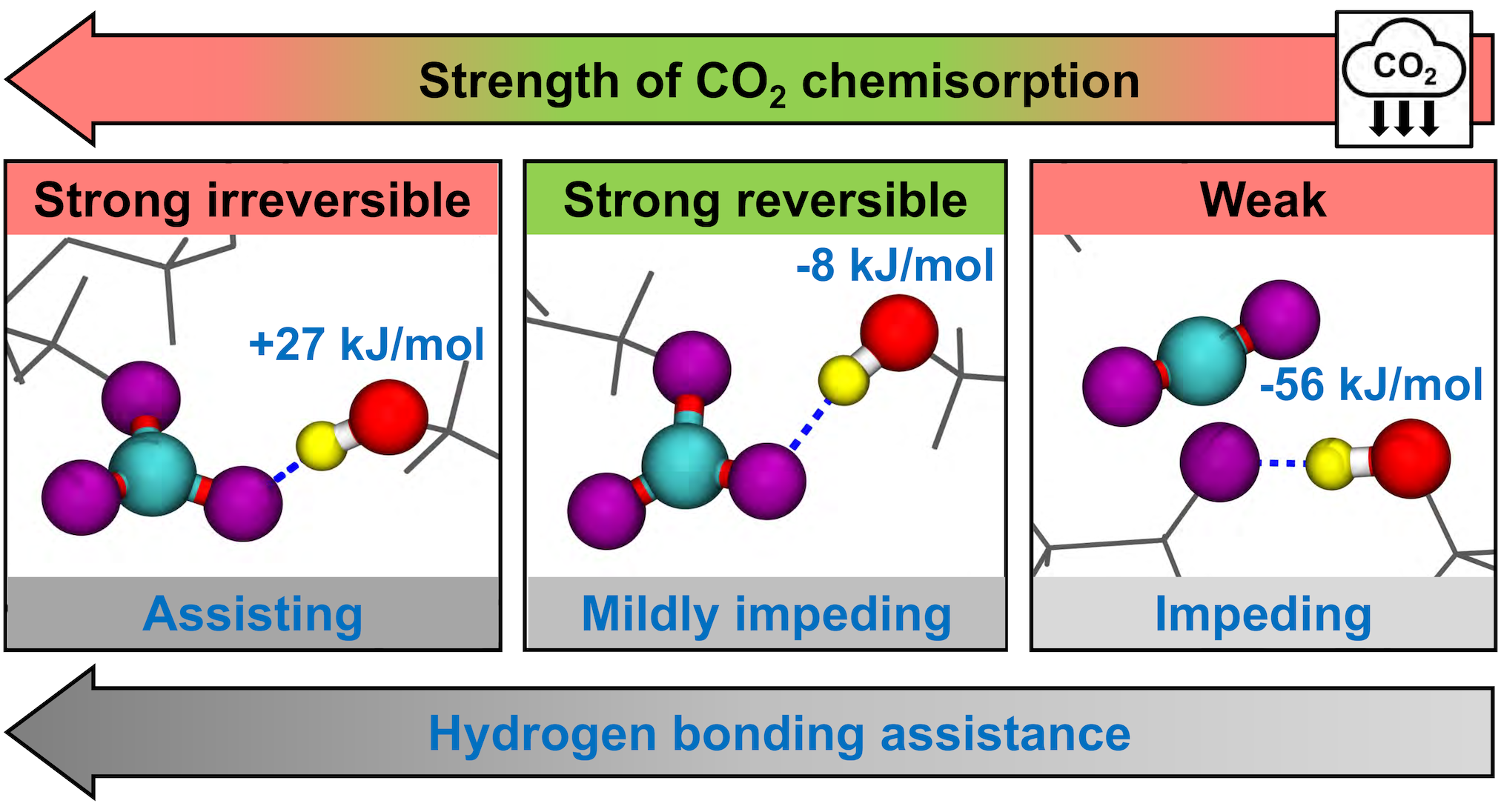
Cyclodextrin-derived metal-organic frameworks (MOFs) have received substantial attention not only because of their remarkable ability to absorb carbon dioxide strongly and reversibly but also because they can be readily obtained from inexpensive, renewable, and environmentally benign components. In our recently completed work, a comprehensive analysis of available experimental data and results of our DFT modeling allowed to reveal the mechanism of the formation of CO2 adsorption sites from inert alcohol groups in these MOFs and to explain why some of the sites bind carbon dioxide with the optimal strength while others bind it too strongly or too weakly. The insights obtained in this work suggest new strategies for advancing design of solid-state materials for CO2 capture or detection.
4. Role of adatoms in the surface-confined Ullmann coupling
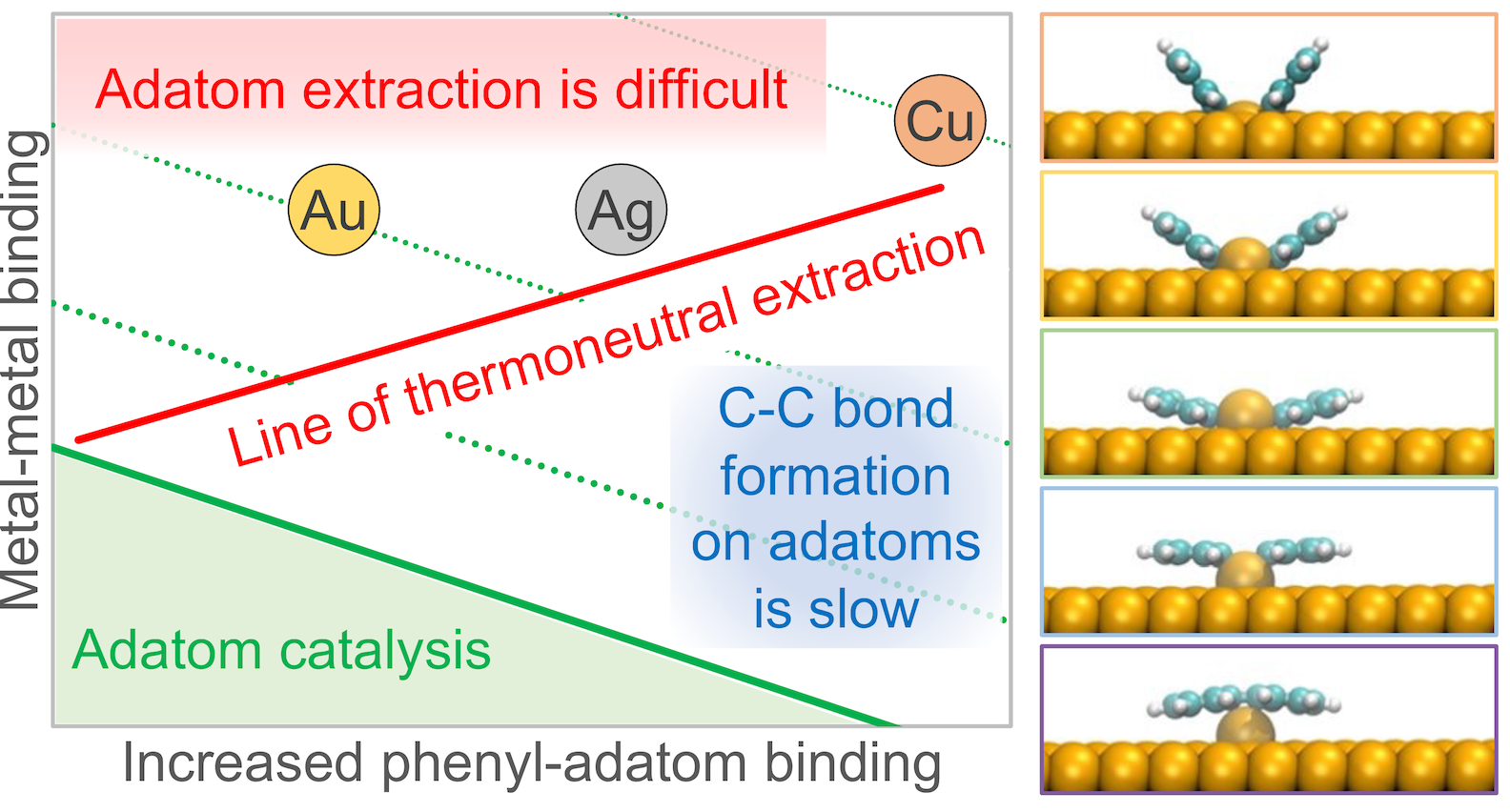
The Ullmann coupling of aromatic molecules on metal surfaces is currently the most successful strategy to synthesize π-conjugated organic polymers with unique electronic properties such as molecular wires, graphene nanoribbons and covalent organic frameworks. In this work, we performed extensive DFT modeling of the adatom creation and adatom-catalyzed Ullmann coupling of monohalogenated benzene molecules on the surfaces of copper, silver and gold with the aim to quantify the potential impact these poorly understood elementary processes may have on the reaction. Our comparative analysis of multiple competing pathways explained why extracted adatoms are difficult to observe during the Ullmann reactions despite the surprising ease of their formation, facilitated by organic intermediates. Our work also demonstrated how the presence of adatoms can lead to the formation of defects in the π-conjugated nanostructures and what metal surfaces can minimize this detrimental process.

Combinatorial study of the Li-La-Zr-O system
Lithium-stuffed garnets based on Li7La3Zr2O12 (LLZO) have attracted a lot of attention as candidate electrolytes for all-solid lithium batteries. Doping…

Correlated local fluctuations in the hydrogen bond network of liquid water
The hypothesis that liquid water can separate into two phases in the supercooled state has been supported by recent experimental…

Light-driven transition-metal-free direct decarbonylation of unstrained diaryl ketones via a dual C–C bond cleavage
The cleavage and formation of carbon−carbon bonds have emerged as powerful tools for structural modifications in organic synthesis. Although transition−metal−catalyzed…

Adatoms in the surface-confined Ullmann coupling of phenyl groups
The on-surface Ullmann coupling of aromatic molecules has emerged as the most successful approach to synthesize atomically precise carbon nanostructures…